With the development of molecular biology and bioinformatics, there is a growing need for a new platform to complete a one-stop platform for analysis, retrieval, design and synthesis
ProbeDesigner is based on this background was born, do not need too deep professional background knowledge, you can complete very professional primer and probe design work
The current software update version of V1.0, follow-up will introduce more features
Contact us:lxk#yhkodia.com
ProbeDesignerSoftware base on BioPython libs, which relies on the Python language, can easily implement regular sequential reads, writes, Blast comparisons, homology analysis, and interaction with Entrez databases, SwissPort processing, Medline database, graphics model, genetic analysis, protein sequence prediction and other functions.
QuantiMATTechnology is a branch-based signal amplification technology based on the development of the second generation of domestic signal amplification technology,ProbeDesignerhe various QuantiMAT experiments can be generated based on the sequence information of the selected target (ML), LE (Label Extender), BL (Blocker). These probe groups passed the severe NSH (nonspecific hybridization analysis), making the probe and universal 2.0 sequence produce the smallest NSH ,To ensure that the experimental high specificity, high sensitivity.
Filtersis a powerful tool for changing the type of probe that allows the service. Using the filter guidelines, you can increase or decrease the number of probes that can be used to detect CES or LES.
It should be noted that the main reason for changing the filtering criteria is to increase the number of CES and LES that can be used to detect. While the default thresholds are excellent guidelines, they are. In many cases, you may have probes that are slightly above or below the allowed threshold.
You, as a probesets designer (I bet you never call), will need to examine the probe in each probe group to determine if any of the features can be easily but does not affect the detection performance.
Keep in mind that the "filter" action (ie, relax the filter standard) relaxes all the probes in the tree. You can not relax the filter for a single probe. When you relax the filter for a probe, you can change the state of the other probes in an advantageous way.
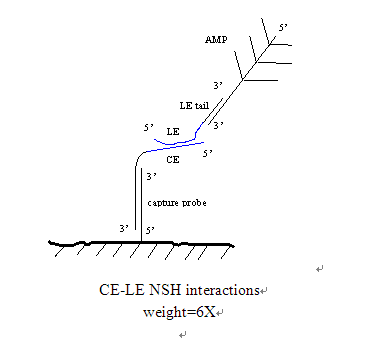
CE-LE interaction known by the ProbeDesigner software is the ultimate source of the background, in fact, these interactions are used to establish the final distribution of the probes in the analysis. Complementary CES and LEs may interact through the following mechanisms. This can be prevented from the service by excluding the same service from one or two probes in the probe group. The method of calculating the probe, calculating the ce-le interaction, and reassigning is necessary, you can imagine an iterative process. Use the assignment view to enable this procedure.
The CE-LE and NSH-calculated ce-le interactions were evaluated using the X-mer algorithm. PD searches for all possible extension primers in the form of x-mer (CEs vs. LEs), estimates their stability, and they are. The same combination of calculations, it will be a heavy X-mer results of the estimated ce-le cross on the background. All ce-le interactions give a weight 6 similar to the le-amp cross-reaction for system 1 detection.
ProbeDesigner which the core of the software is the x-mer algorithm. The algorithm is used to assess the hybridization stability between any universal sequence
"X-mer" refers to the length of the perfect perfect hybrid determined by the algorithm. Although the default value of the program is a 4-mer, you can adjust this parameter.When the algorithm: The x-mer algorithm begins to identify all possible x-mers between the selected probe and the generic sequence, a simple weighting scheme is used to predict every perfect,X-mer mixed stability, and summarizes the cross-reactivity of each probe with the universal sequence for all hybridization results. The following example:
1. All possible 4-mers are defined as follows:
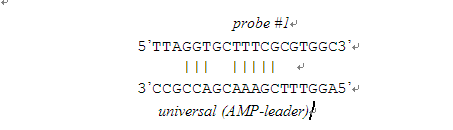
There are two 4-mers: TTTC-GAAA and TTCG-CGAA
2. Any x-mer (with a typical 4-mers as an example) is calculated by passing a simple weighting scheme.
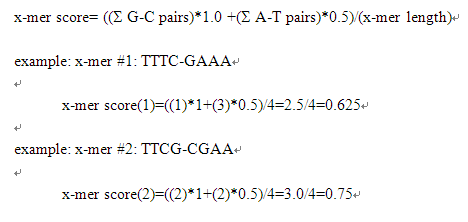
And 3. all of the 4-mers calculated fractional multi-universal probe sequences and design probes cross-reactive factor values
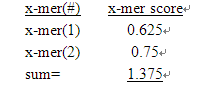
Once the x-mer algorithm produces the result of the cross-reactivity of all possible general sequences, the NSH algorithm calls.
1. Each x-mer score is multiplied by the corresponding weighting factor (1X CE with AP, 6X CE amplifier leading, 15 times CE and arm) to determine the NSH score of the probe. There is a result of the cross-reactivity of each probe with the universal sequence. This process is performed once assuming that the probe is a CE, once assumed it is a LE. In this example, suppose we are looking for a cross-reaction of ce-amp with the designed probe
NSH Score = x-mer values * weight factor = 1.375 * 6 =8.25
2. The fraction of the x-mer is calculated to interact with the other probe NSH (which means that the x-mer algorithm also recalculates the cross-reactivity between the probes)。

NSH defination
The NSH response involves the interaction of the capture primer set and the marker extension primer set and the universal sequence leads to a high detection background in the bDNA assay..Minimizing the NSH response is an essential part of a successful primer set design. Therefore, Probedesigner is able to screen out primer probes to exclude those that may cause background elevation through NSH reactions. (Citing a literature on NSH responses,see M. Collins et. al., NAR Vol 25, no. 15, Aug 1997).
Before you start, an important thing is how we define a "generic" sequence. These molecules, which are common in all BDNA detection systems (ie, the same general chemical type), mediate target capture and signal amplification For the SYSTEM 1 detection system (open kit), they included branching DNA molecules, AP probes, and capture probes (PSCP) on the coated plate. For the SYSTEM 8 detection system, they included branching DNA molecules (with iso-C, iso-G), preloaded macromolecules and capture probes on the plate, CE The tail of the CE and LE primer sets (also extended) are also considered generic, and after splicing with specific extension sequences, they co-ordinate the possibility that the probes are hybridized to the universal sequence and subsequent signal amplification is possible. For any chemical system, these generic sequences are combined together to form a file into the ProbeDesigner software to become a group
-
Screening a probe from a probe set may result in a high background involving the following aspects:
- Identify a general extension that can lead to nonspecific hybridization;
- Evaluate the hybrid stability that is formed between the universal and extended sequences;
- Evaluate the impact of any potential stable cross reaction on the background.
For the NSH cross reaction System 1 chemical system is described so. The cross-reactivity varies depending on whether the primer is as a CE primer or a LE primer. In the early design process, we did not specify the type of primers, and any primers could be CE primers or LE primers. Thus, for all primer sets are two analyzes, one hypothesis as CE primitive service, one hypothesis as LE primer service.
Probedesigner estimates hybridization stability and its background effects. The interaction stability of each interaction is calculated using the X-ray algorithm (discussed below), and the contribution of the mixed background is multiplied by the stability of this predicted weighting factor. The common score for each interaction-related system 1 is as follows. Although it is recommended to use the default settings, the mix parameters and the NSH weighting factor can be changed within the program using the NSH dialog box.
As an example, note that the ce-amp arm interacts 15 times more. If the stability of the mixture is calculated from the X-mer algrithm, say "1", this interaction with the CE NSH score is 15 points (15 times the result of the Fulun machinery manufacturing). Now, if you are concerned, you want to know why CE - "amplifier - ARM" interaction gives more weight than the chief executive "AMP Leader" and the Chief Executive - "AP Probe" interaction. For the 15x3 molecule, each AMP weapon has an AP probe triple binding site. This would result in 45 binding sites for AP and CE probes that should have nonspecific pairing. Conceptually you can imagine why this is an interaction and we want to avoid NSH.
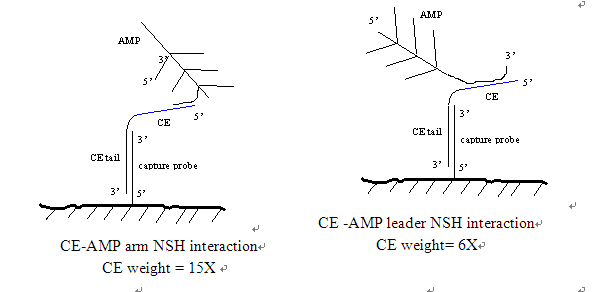
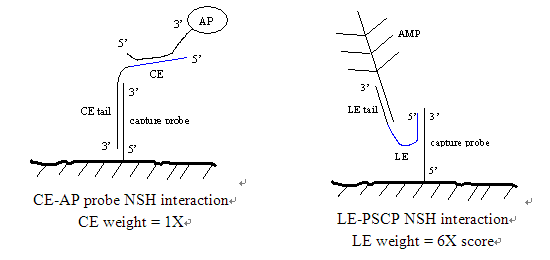
The NSH cross reaction for the System 1 chemical system